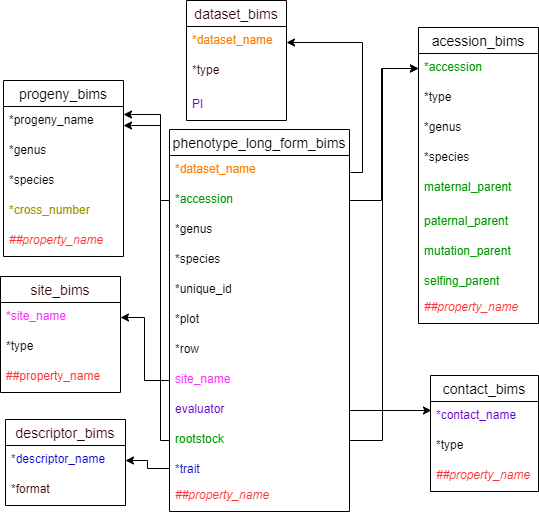
Phenotype template
There are two templates that can be used to enter phenotype data. Two templates are the same except that descriptors are entered as column headings in phenotype_bims and as data in the trait column in phenotype_long_form_bims. As a result, there will be one row per each sample for phenotyping in phenotype_bims and there will be multiple rows per each sample for phenotyping in phenotype_long_form_bims as shown in the diagram below. As shown below, the descriptors in phenotype_bims template should have # prefix.
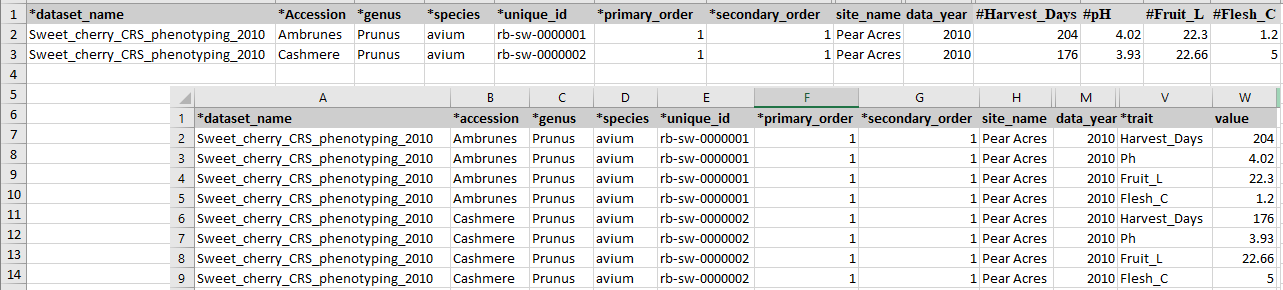
As explained in the Creating a New Breeding Program section, the names of the four columns, accession, unique_id, primary_order, and secondary_order, can be changed and the downloaded template will reflect the change. The unique_id represents a unique phenotyping sample and it should be unique within the dataset name. The columns primary_order and secondary_order are for the plot design such as plot and row. If those are not relevant for the program, breeders can just add the same number for each row as shown in the figure above. If there are any data for specific samples, such as specific sample treatment, users can specify it in the property_bims and use it as a column heading with ## as a prefix.
Below are descriptions for each of the columns in phenotype_bims. Columns with * are required.
- * dataset_name: name of the phenotyping dataset. It should match a 'dataset_name' column entry in the dataset_bims.
- * accession: ID of the accession that has been phenotyped. It should match an 'accession' column entry in accession_bims or ‘progeny_name' column entry in progeny_bims.
- * genus: genus to which the accession belongs to.
- * species: species name. Enter 'sp.' to represent one unknown species, 'spp.' to represent multiple unknown species.
- * unique_id: Unique ID of the sample. It should be unique within the dataset.
- * primary_order: The primary order of a sample
- * secondary_order: The secondary order of a sample
- clone_ID: ID of a specific clone if available (e.g. individual tree).
- evaluator: person who did the phenotyping. Multiple persons can be entered with ';' in between. It should match the contact_name of contact_bims.
- site_name: site information where the accession for the phenotyping is planted. It should match 'site_name' in the site_bims.
- rootstock: name of the rootstock in tree breeding program if the scion is being phenotyped. It should match an 'accession' column of accession_bims.
- plant_date: date of the planting
- data_year: phenotyping year if only year is known
- evaluation_date: date of phenotype evaluation
- pick_date: date of the sample collection if the collection is done on a different date than the phenotype evaluation.
- previous_entry: accession of the previous entry if the name of the accession has been changed after initial trials.
- barcode: barcode
- fiber_pkg: group of samples for phenotyping, can contain samples from multiple germplasm (used mainly by cotton breeders).
- storage_time: time between collection and phenotyping.
- storage_regime: the condition of sample storage between the collection and phenotyping.
- comments: any comments for the phenotyping.
- #descriptor_name: special columns (#) : The name of the marker is entered as a column heading with ‘#’ as a prefix. It should match the ‘descriptor_name’ column entry in the descriptor_bims. The phenotypic values are entered in the cell.
- ##property_name: user specified custom columns that should have ## as a prefix. It should match an entry in propery_bims with the type specified as phenotype
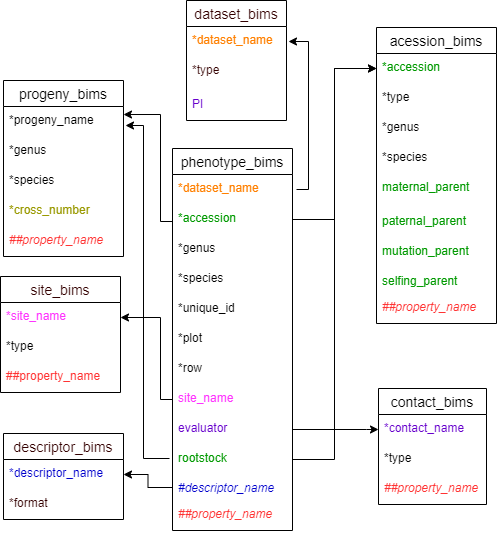
Diagram below shows the diagram of phenotype_bims along with the other templates where the columns in phenotype_bims should match. Accession and rootstock can match either accession in accession_bims or progeny_name in probeny_bims. Descritor_names in descriptor_bims can be used as column headings in phenotype_bims with # prefix.
Below are descriptions for each of the columns in phenotype_long_form_bims. Columns with * are required.
- * dataset_name: name of the phenotyping dataset. It should match a 'dataset_name' column entry in the dataset_bims.
- * accession: ID of the accession that has been phenotyped. It should match an 'accession' column entry in accession_bims or ‘progeny_name' column entry in progeny_bims.
- * genus: genus to which the accession belongs to.
- * species: species name. Enter 'sp.' to represent one unknown species, 'spp.' to represent multiple unknown species.
- * unique_id: Unique ID of the sample. It should be unique within the dataset.
- * primary_order: The primary order of a sample
- * secondary_order: The secondary order of a sample
- clone_ID: ID of a specific clone if available (e.g. individual tree).
- evaluator: person who did the phenotyping. Multiple persons can be entered with ';' in between. It should match the contact_name of contact_bims.
- site_name: site information where the accession for the phenotyping is planted. It should match 'site_name' in the site_bims.
- rootstock: name of the rootstock in tree breeding program if the scion is being phenotyped. It should match an 'accession' column of accession_bims.
- plant_date: date of the planting
- data_year: phenotyping year if only year is known
- evaluation_date: date of phenotype evaluation
- pick_date: date of the sample collection if the collection is done on a different date than the phenotype evaluation.
- previous_entry: accession of the previous entry if the name of the accession has been changed after initial trials.
- barcode: barcode
- fiber_pkg: group of samples for phenotyping, can contain samples from multiple germplasm (used mainly by cotton breeders).
- storage_time: time between collection and phenotyping.
- storage_regime: the condition of sample storage between the collection and phenotyping.
- comments: any comments for the phenotyping.
- *trait: name of the trait descriptor
- value: The phenotypic value
- ##property_name: user specified custom columns that should have ## as a prefix. It should match an entry in propery_bims with the type specified as phenotype
Diagram below shows the phenotype_long_form_bims template along with the other templates where the columns in phenotype_long_form_bims should match. Accession and rootstock can match either accession in accession_bims or progeny_name in probeny_bims. Descritor_names in descriptor_bims can be used as data in the trait column.