Entering Data into Templates
Entering Data into Templates sookEntering Data into Templates
This section describes in detail how to enter data in BIMS templates.
Template Overview
Template Overview sookTemplate Overview
To import data into BIMS, the data needs to be in the BIMS templates. Phenotype data can also be imported to BIMS using exported files from Field Book or BrAPI, but data other than phenotype, such as contact, dataset, site, cross, progeny, and accession, need to be loaded using templates.
Data templates are available for download in the Template list under the Data Import section on the left-hand accordion menu. In the Data Templates tab, users can view the description of each template and download the templates. Data templates are Excel files that contain metadata types as headings where users can enter data to be loaded into the database. In the current version of BIMS, templates are available for each of the following data types: property, contact, dataset, trait descriptor, site, accession, cross, progeny, marker, haplotype block, phenotype, genotype, haplotype, and image. There can be multiple templates for some data types.
As explained in Creating a New Breeding Program section of the BIMS manual, users can choose to change the names of the required column names for Field Book App and the phenotype templates (phenotype_long_form_bims or phenotype_bims) of the template. This can be done when users create a new breeding program or in the BIMS/Field Book section of the configuration panel shown below. The change made here will be reflected in the template that the user downloads.
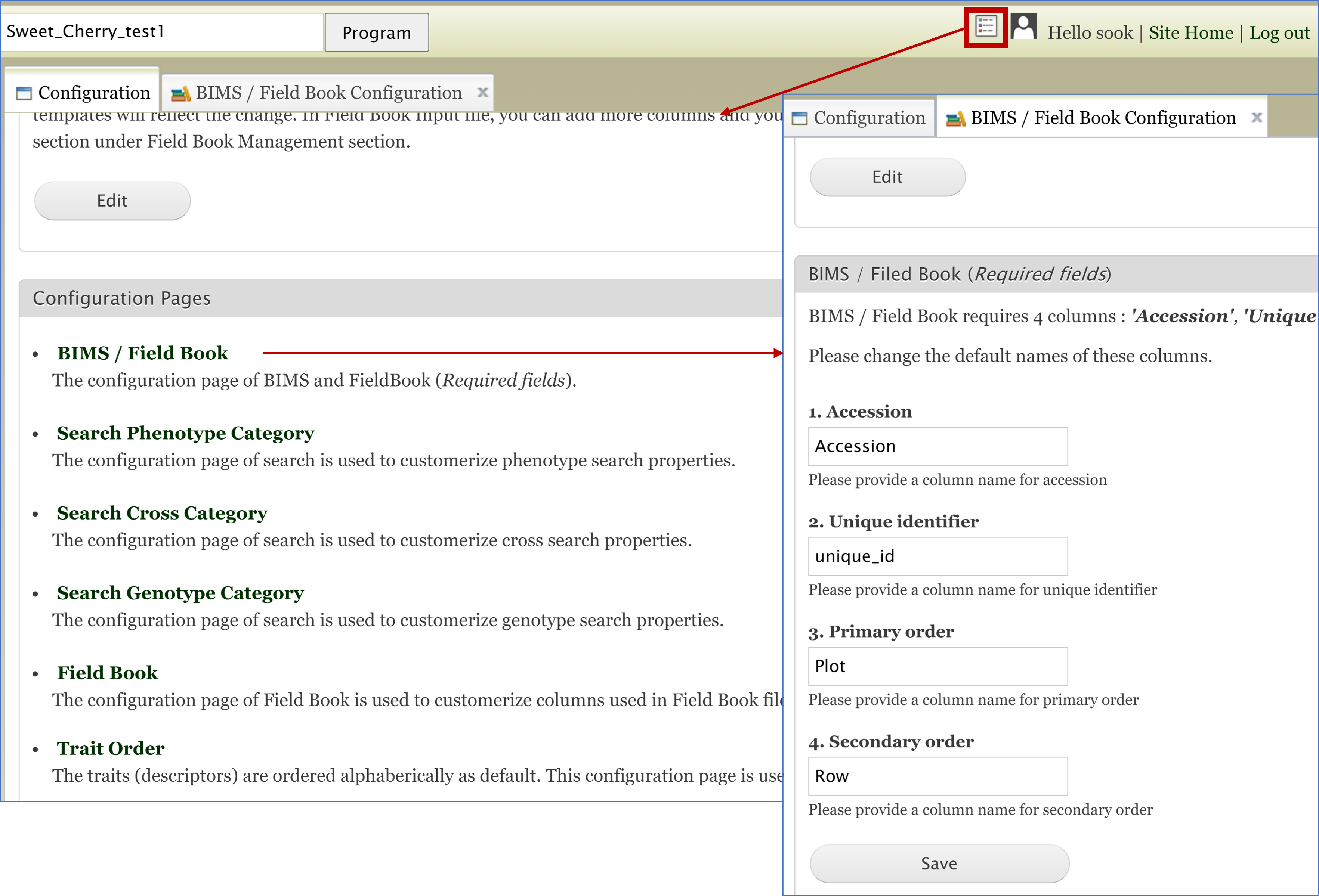
The same names of the four required columns should be used when preparing an input file for Field Book App. The figure below shows matching column names in the Field Book App input file and phenotype_bims template.
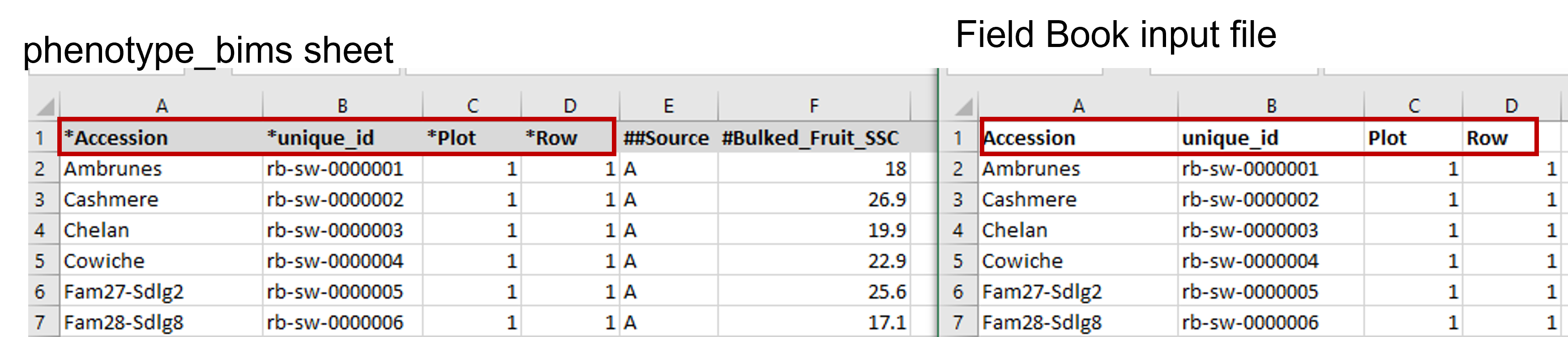
Different types of columns
In each template, the required columns have * as a prefix as shown above. Other columns can be used to enter data or left empty. Columns that are not specified in the templates will be ignored when loading data. Users can, however, make custom columns in some of the templates which should be specified in the property_bims and have ## as a prefix of the column name as shown above. In phenotype_bims, trait descriptor names are entered as column headings, and they need to have # as a prefix as shown above. The trait descriptor names need to match an entry in the descriptor_name column in the descriptor_bims if they have not been loaded to BIMS already. In the genotype_snp_wide_form_bims, marker names are entered as column headings, and they need to have $ as a prefix. The marker names need to match marker_name in the marker_bims if they have not been loaded to BIMS already. For some columns, only one of the specified terms can be entered as valid data. For example, valid entries for the type column in contact_bims are person, company, database, institution, lab, and organization. Those columns and their valid entries will be described in each data template section.
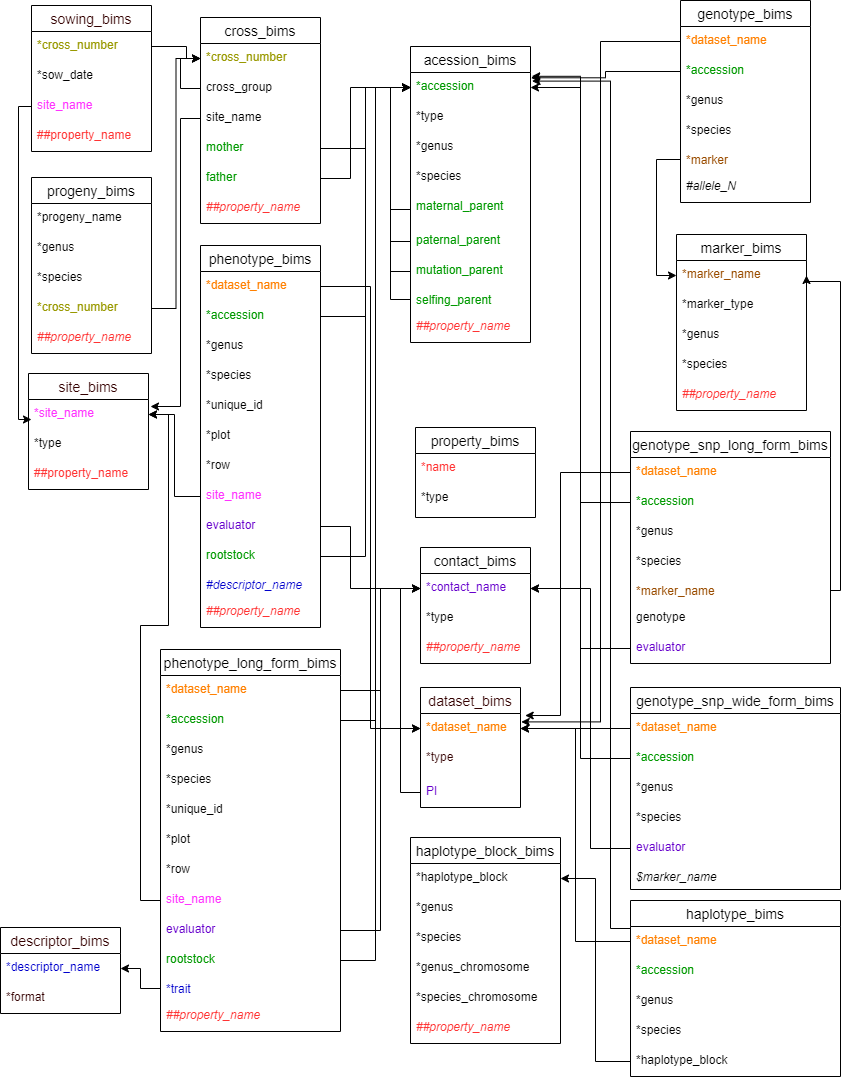
BIMS template diagram
BIMS template diagram sookBIMS template diagram
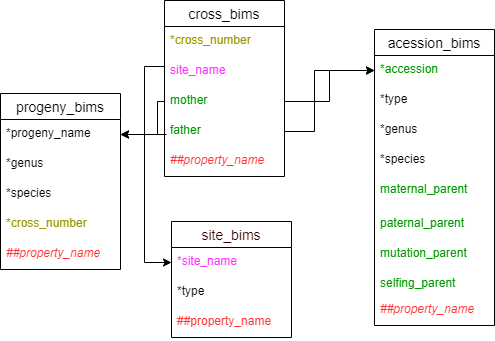
Data in some of the columns refer to data in other templates or data already entered to BIMS so the data needs to match exactly. For example, phenotype_bims has the dataset_name column and the data entry should match the data in the dataset_name column of the dataset_bims. Once an entry in the dataset_bims is loaded to BIMS or the dataset names are entered using the BIMS interface, users can enter it in phenotype_bims without entering the same entry again in dataset_bims. The data types that need to be entered to BIMS either using the template or the BIMS interface before being used in other templates include dataset, accession, trait descriptor, and site. Contact, custom property, marker, and haplotype block data can also be used in other templates and their detailed data need to be entered to BIMS using contact_bims, property_bims, marker_bims, and haplotype_bims. Entries in property_bims, descriptor_bims, and marker_bims can be used as column headings in some of the other templates and they need to have a special prefix when used as headings; # for descriptor_name, ## for property_name, and $ for marker_name.
Figure below shows all the templates available in BIMS with column names that are either required or have relationships with other columns. Matching column names have the same color and they are connected by an arrow. Arrows between property_name columns were omitted to make the diagram less busy. Progeny_name in progeny_bims can also be used, as well as accession in accession_bims, in entering accession information in other templates. However the arrows to the progeny_name in progeny_bims were skipped to make the diagram simpler. Matching columns in various templates do not always have the same column headings. For example, the evaluator column in phenotype_bims refers to a contact_name in contact_bims.
Property template
Property template sookProperty template
Users can make custom columns in some of the templates and those custom columns should be specified in the property_bims. Templates that can have custom columns include contact_bims, cross_bims, sowing_bims, site_bims, accession_bims, progeny_bims, marker_bims, haplotype_block_bims, phenotype_bims, genotype_snp_wide_form_bims, haplotype_block_bims, and marker_bims.
Below are descriptions for each of the columns in property_bims. Columns with * are required. Type is a validated column.
- *name: name of the property
- *type: type of the property. Valid data entries are contact, cross, sowing, site, accession, progeny, marker, haplotype_block, phenotype, snp, and haplotype_block.
- alias: alias for the property
- Definition: definition of the custom property
Different types of columns
In each template, the required columns have * as a prefix as shown above. Other columns can be used to enter data or left empty. Columns that are not specified in the templates will be ignored when loading data. Users can, however, make custom columns in some of the templates which should be specified in the property_bims and have ## as a prefix of the column name as shown above. In phenotype_bims, trait descriptor names are entered as column headings, and they need to have # as a prefix as shown above. The trait descriptor names need to match an entry in the descriptor_name column in the descriptor_bims if they have not been loaded to BIMS already. In the genotype_snp_wide_form_bims, marker names are entered as column headings, and they need to have $ as a prefix. The marker names need to match marker_name in the marker_bims if they have not been loaded to BIMS already. For some columns, only one of the specified terms can be entered as valid data. For example, valid entries for the type column in contact_bims are person, company, database, institution, lab, and organization. Those columns and their valid entries will be described in each data template section
Dataset template
Dataset template sookDataset template
Users can define a dataset in any way they want to, but a dataset in breeding data typically refers to a trial. For phenotyping data, a set of data can belong to the same dataset as long as a unique ID in the phenotype templates gives a single phenotype value per descriptor. For users who use the Field Book App for phenotype collection, a single input file for the app, with a unique ID in each row, typically corresponds to a single dataset. Since only one phenotypic value can be given to the same unique ID in the Field Book App input file, users can decide to make two different input files and save them as two different datasets to record phenotypic values collected by two different people. For genotyping data, a set of data can belong to the same dataset as long as an accession gives a single genotype per marker. The dataset template can be filled with other templates, and all the template sheets can be put in the same Excel file and uploaded together.
Below are descriptions for each of the columns in dataset_bims. Columns with * are required. Type and sub_type are validated columns.
- *dataset_name: name of the dataset
- *type: type of the dataset. Valid data entries are phenotype and genotype.
- sub_type: subtype of the dataset. Valid subtype of genotype dataset includes SSR, SNP and haplotype.
- description: general description of the dataset
- PI: main breeder of the program or principal Investigator of the dataset for collaborative projects. It should match an entry in the contact_name column of the contact_bims.
- ##property_name: user specified custom columns that should have ## as a prefix. It should match an entry in propery_bims with the type specified as dataset.
Contact template
Contact template sookContact template
Contact_bims template is used to enter data related to a person or an institution. The contact entry can then be used in other templates such as phenotype_bims and accession_bims. In phenotype_bims, for example, there is a column called collector and users can enter one of the entries in contact_bims. In accession_bims, there is a column called germplasm_center and users can use one of the entries in contact_bims.
Below are descriptions for each of the columns in contact_bims. Columns with * are required. Type is a validated column.
- * contact_name: name of the contact. For contact_name of a person, it is recommended to enter full name (eg. John M. Smith).
- alias: any alias of the contact
- * type: type of the contact. Valid entries are person, company, database, institution, lab, and organization.
- first_name: First name of the person
- last_name: Last name of the person
- ORCID: ORCID (Open Researcher and Contributor ID) which is a nonproprietary alphanumeric code to uniquely identify scientific and other academic authors and contributors.
- institution: name of the institution the person belongs to. This doesn’t have to be a separate entry in contact_bims.
- lab: name of the lab the person belongs to. This doesn’t have to be a separate entry in contact_bims.
- address: address of the contact
- email: email address of the contact
- phone: phone number of the contact
- fax: fax number of the contact
- title: job title of the person
- state: state of the contact
- country: country of the contact
- source: source of the data when the contact information is obtained from other databases
- last_update: date of the last update
- url: URL of homepage of the contact
- comments: any comments of the contact
- keywords: a maximum of 5 keywords that best describe the person's area of work
- ##property_name: user specified custom columns that should have ## as a prefix. It should match an entry in propery_bims with the type specified as contact.
Site template
Site template sookSite template
Site_bims is used to collect the data of the location of the crop.
Below are descriptions for each of the columns in site_bims. Columns with * are required. Type is a validated column.
- * site_name: name of the site
- site_long_name: long name of the site.
- * type: type of the location. Valid entries are greenhouse, open field, orchard, seedling block, and retail.
- latitude : the decimal latitude coordinate of the georeference, using positive and negative signs to indicate N and S, respectively.
- longitude: the decimal longitude coordinate of the georeference, using positive and negative signs to indicate E and W, respectively.
- altitude : the altitude (elevation) of the location in meters. If the altitude is only known as a range, enter the average.
- geodetic_datum: the geodetic system on which the geo-reference coordinates are based. For geo-references measured between 1984 and 2010, this will typically be WGS84.
- country: country of the location
- state: state of the location
- region: region of the location
- address: the entire address except the country
- comments: any further comments on the site
- ##property_name: user specified custom columns that should have ## as a prefix. It should match an entry in propery_bims with the type specified as site.
Descriptor template
Descriptor template sookDescriptor template
Descriptor_bims is used to enter trait descriptors used in phenotyping. To support the use of Field Book App, the format of the trait descriptors are validated to be the same as those in Field Book App.
Below are descriptions for each of the columns in descriptor_bims. Columns with * are required. Format is a validated column.
- *descriptor_name: any trait descriptor that a breeder uses in their own program. These descriptors can be used as column heads in phenotype_bims, with a '#' prefix.
- alias: alias of the descriptor.
- *format: The format of the descriptor. Valid entries are audio, boolean, categorical, counter, date, disease_rating, location, multicat, numeric, percent, photo, and text.
- valid format for date type: d-m-yyyy, dd-mm-yyyy, yyyy-m-d, yyyy-mm-dd, d/m/yyyy, dd/mm/yyyy, yyyy/m/d, or yyyy/mm/dd. Note year should be in four digits.
- trait_name: Breeders normally leave this column empty. This is only to specify crop trait ontology terms for specific reasons.
- categories: The values of the categorical descriptor separated by '/' (eg. red/white/pink). Only the categories described here can be entered into BIMS as valid values of categorical descriptors used in phenotype_bims and phenotype_long_form_bims.
- data_unit: unit for the trait descriptor
- minimum: the minimum value of the descriptor
- maximum: the maximum value of the descriptor
- defaultValue: the default value of the descriptor
- definition: definition of the descriptor
Accession template
Accession template sookAccession template
Accession_bims is used to enter data of accessions in the program. As described in the Template Overview section of the BIMS manual, if users change ‘accession’ to something else (i.e. stock), the names of the tab and the column downloaded in their BIMS program will reflect that change. For example if they change accession to stock, the name of the tab will change from accession_bims to stock_bims and the name of the column will change from accession to stock.
Accessions are subjects that are used in phenotyping, genotyping, and crossing. This data can be entered either in accession_bims or progeny_bims. As shown below, if the accession is from a specific cross and it has associated cross information, the data can be entered in progeny_bims. Otherwise the accession data can be entered in accession_bims.
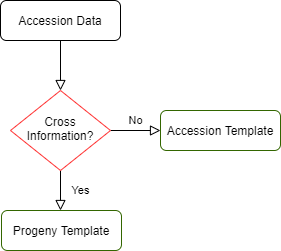
Below are descriptions for each of the columns in accession_bims. Columns with * are required. Type is a validated column.
- * accession: name of accession. The most important ID or name (unique and stable at least within the breeding program) should be in this column.
- * type: type of the accession. Valid options are cultivar, breeding_research_material, and wild_unimproved for an individual accession. Population can be used for a group of individuals.
- * genus: genus of the accession
- * species: species name of the accession. Enter ‘sp.’ when the actual species name cannot or need not be specified. Enter the plural form ‘spp.’ when referring to several species within the genus. Please check if your species exists in our database by going to Data Import-Data Lookup-Organisms in BIMS. If they do not exist, please contact us.
- GRIN_ID: Accession ID (ACID) in the GRIN database, if it is known. Multiple GRIN ID, separated by comma, can be added.
- subspecies: subspecies name.
- secondary_ID: alternate ID or name that is commonly used to refer to the entry. This represents an alias of importance and it does not have to be entered in the alias column separately.
- maternal_parent: accession name of the maternal parent. This needs to be entered as a separate row in accession_bims.
- paternal_parent: accession name of the paternal parent. This needs to be entered as a separate row in accession_bims.
- mutation_parent: name of the accession used in the mutant breeding. This needs to be entered as a separate row in accession_bims.
- selfing_parent: name of the accession used in self-fertilization. This needs to be entered as a separate row in accession_bims.
- alias: a concatenation of all other less commonly used names or designations for the entry. The format is 'Alias type1:Alias1; Alias type2:Alias2' (e.g. Collector:98HT-227; Site:W6 21306). When there is no specific type for aliases, just write the aliases without the specific type (e.g. ABC-1; 21306).
- cultivar: cultivar name if It is a named cultivated variety. It can be the same as the accession name.
- pedigree: any additional pedigree information when the exact parents are not known, or any additional information beyond just parents (e.g. Red-fruit sport of Jonathan, Pyrus sp. X Cydonia sp., etc).
- origin_detail: detailed description of the origin of the accession
- origin_country The original country for the variety (especially for wild variety).
- population_size: only for type 'population'.
- germplasm_center: germplasm center or lab where the accession is distributed.
- description: any description for the accession
- comments: any comments on the accession
- ##property_name: user specified custom columns that should have ## as a prefix. It should match an entry in propery_bims with the type specified as accession.
Cross template
Cross template sookCross template
Cross_bims is used to enter data related to a cross that is performed in the breeding program. Users can choose to treat cross_number as a specific parental set, or a specific cross done on a certain date.
Below are descriptions for each of the columns in cross_bims. Columns with * are required.
- site_name: site information where the cross was done. It should match an entry in the site_name column of the site_bims template.
- * cross_number: ID number or name for a specific cross.
- mother : name of the seed parent for the cross. It should match an entry in the accession_name column of the accession template or progeny_name in the progeny template.
- mother_location: location details for the mother of this cross
- father: name of the pollen parent for the cross. It should match an entry in the accession_name column of the accession_bims or progeny_name in the progeny_bims.
- father_location: location details for the father of this cross
- cross_date: date for the cross
- comments: any comments on the cross
- ##property_name: user specified custom columns that should have ## as a prefix. It should match an entry in propery_bims with the type specified as cross.
Package template
Package template sookPackage template
Package_bims is used to keep track of the number of seeds that are sown. Breeders can sow seeds from the same cross multiple times and this template can be used to record how many seeds have been sown at a particular date. Seeds from the same cross may be put into one or multiple seed packages.
Below are descriptions for each of the columns in sowing_bims. Columns with * are required.
- *package_number: ID number or name of a seed package
- cross_number: ID number or name of a cross where the seed came from. It should match an entry in the cross_number column of the cross_bims template.
- cross_date: when the cross_number represents a parental set that have been done multiple times, a specific cross date for the package can be entered
- * sow_date: The date of the sowing
- num_seeds_sown: the number of the seeds sown
- notes: Any notes on the sowing
- ##property_name: user specified custom columns that should have ## as a prefix. It should match an entry in propery_bims with the type specified as sowing.
Progeny template
Progeny template sookProgeny template
Progeny_bims is used to enter accession data with cross information. The accession data that are entered in the progeny_bims do not need to be entered again in accession_bims, and the entry in progeny_name can be used in accession or rootstock columns in other templates.
Below are descriptions for each of the columns in progeny_bims. Columns with * are required.
- * progeny_name: name of the progeny
- alias: Multiple aliases of the progeny can be entered here. The format is 'alias type1:alias1; alias type2:alias2'. When there is no specific type for an alias, just write the aliases without the specific type.
- * genus: genus of the progeny
- * species: species name of the progeny. Enter ‘sp.’ when the actual species name cannot or need not be specified. Enter the plural form ‘spp.’ when referring to several species within the genus.
- * cross_number: Specific ID of a cross that generated the progeny; Must also be found in the cross_bims template.
- description: Any other descriptive data about the selection or seedling.
- comments: Comments on the progeny.
- advanced_to_2nd: Yes if it is advanced to 2nd phase, No otherwise. Leave it empty if the selection decision is yet to be made.
- advanced_to_3rd: Yes if it is advanced to 3rd phase, No otherwise.
- advanced_to_4th: Yes if it is advanced to 4th phase, No otherwise.
- ##property_name: user specified custom columns that should have ## as a prefix. It should match an entry in propery_bims.
Phenotype template
Phenotype template sookPhenotype template
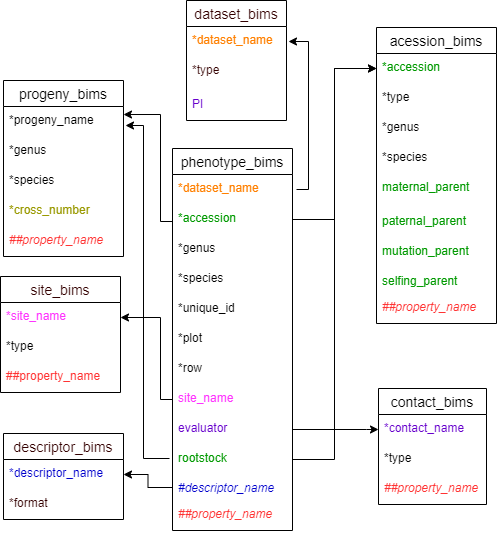
There are two templates that can be used to enter phenotype data. Two templates are the same except that descriptors are entered as column headings in phenotype_bims and as data in the trait column in phenotype_long_form_bims. As a result, there will be one row per each sample for phenotyping in phenotype_bims and there will be multiple rows per each sample for phenotyping in phenotype_long_form_bims as shown in the diagram below. As shown below, the descriptors in phenotype_bims template should have # prefix.
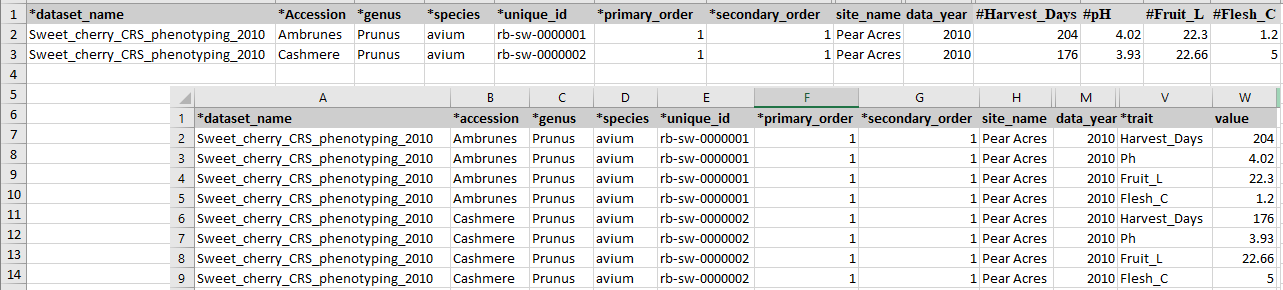
As explained in the Creating a New Breeding Program section, the names of the four columns, accession, unique_id, primary_order, and secondary_order, can be changed and the downloaded template will reflect the change. The unique_id represents a unique phenotyping sample and it should be unique within the dataset name. The columns primary_order and secondary_order are for the plot design such as plot and row. If those are not relevant for the program, breeders can just add the same number for each row as shown in the figure above. If there are any data for specific samples, such as specific sample treatment, users can specify it in the property_bims and use it as a column heading with ## as a prefix.
Below are descriptions for each of the columns in phenotype_bims. Columns with * are required.
- * dataset_name: name of the phenotyping dataset. It should match a 'dataset_name' column entry in the dataset_bims.
- * accession: ID of the accession that has been phenotyped. It should match an 'accession' column entry in accession_bims or ‘progeny_name' column entry in progeny_bims.
- * genus: genus to which the accession belongs to.
- * species: species name. Enter 'sp.' to represent one unknown species, 'spp.' to represent multiple unknown species.
- * unique_id: Unique ID of the sample. It should be unique within the dataset.
- * primary_order: The primary order of a sample
- * secondary_order: The secondary order of a sample
- clone_ID: ID of a specific clone if available (e.g. individual tree).
- evaluator: person who did the phenotyping. Multiple persons can be entered with ';' in between. It should match the contact_name of contact_bims.
- site_name: site information where the accession for the phenotyping is planted. It should match 'site_name' in the site_bims.
- rootstock: name of the rootstock in tree breeding program if the scion is being phenotyped. It should match an 'accession' column of accession_bims.
- plant_date: date of the planting
- data_year: phenotyping year if only year is known
- evaluation_date: date of phenotype evaluation
- pick_date: date of the sample collection if the collection is done on a different date than the phenotype evaluation.
- previous_entry: accession of the previous entry if the name of the accession has been changed after initial trials.
- barcode: barcode
- fiber_pkg: group of samples for phenotyping, can contain samples from multiple germplasm (used mainly by cotton breeders).
- storage_time: time between collection and phenotyping.
- storage_regime: the condition of sample storage between the collection and phenotyping.
- comments: any comments for the phenotyping.
- #descriptor_name: special columns (#) : The name of the marker is entered as a column heading with ‘#’ as a prefix. It should match the ‘descriptor_name’ column entry in the descriptor_bims. The phenotypic values are entered in the cell.
- ##property_name: user specified custom columns that should have ## as a prefix. It should match an entry in propery_bims with the type specified as phenotype
Diagram below shows the diagram of phenotype_bims along with the other templates where the columns in phenotype_bims should match. Accession and rootstock can match either accession in accession_bims or progeny_name in probeny_bims. Descritor_names in descriptor_bims can be used as column headings in phenotype_bims with # prefix.
Below are descriptions for each of the columns in phenotype_long_form_bims. Columns with * are required.
- * dataset_name: name of the phenotyping dataset. It should match a 'dataset_name' column entry in the dataset_bims.
- * accession: ID of the accession that has been phenotyped. It should match an 'accession' column entry in accession_bims or ‘progeny_name' column entry in progeny_bims.
- * genus: genus to which the accession belongs to.
- * species: species name. Enter 'sp.' to represent one unknown species, 'spp.' to represent multiple unknown species.
- * unique_id: Unique ID of the sample. It should be unique within the dataset.
- * primary_order: The primary order of a sample
- * secondary_order: The secondary order of a sample
- clone_ID: ID of a specific clone if available (e.g. individual tree).
- evaluator: person who did the phenotyping. Multiple persons can be entered with ';' in between. It should match the contact_name of contact_bims.
- site_name: site information where the accession for the phenotyping is planted. It should match 'site_name' in the site_bims.
- rootstock: name of the rootstock in tree breeding program if the scion is being phenotyped. It should match an 'accession' column of accession_bims.
- plant_date: date of the planting
- data_year: phenotyping year if only year is known
- evaluation_date: date of phenotype evaluation
- pick_date: date of the sample collection if the collection is done on a different date than the phenotype evaluation.
- previous_entry: accession of the previous entry if the name of the accession has been changed after initial trials.
- barcode: barcode
- fiber_pkg: group of samples for phenotyping, can contain samples from multiple germplasm (used mainly by cotton breeders).
- storage_time: time between collection and phenotyping.
- storage_regime: the condition of sample storage between the collection and phenotyping.
- comments: any comments for the phenotyping.
- *trait: name of the trait descriptor
- value: The phenotypic value
- ##property_name: user specified custom columns that should have ## as a prefix. It should match an entry in propery_bims with the type specified as phenotype
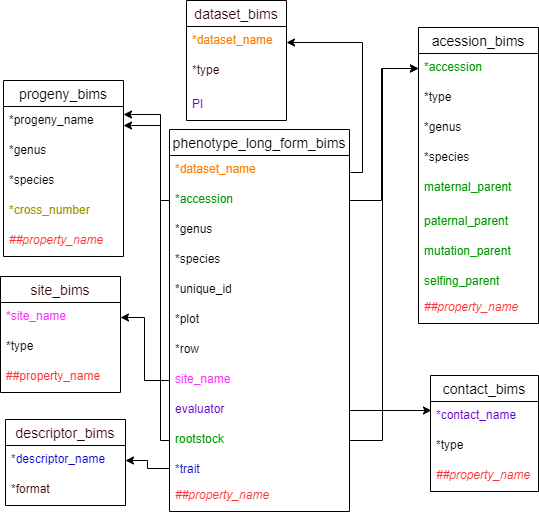
Diagram below shows the phenotype_long_form_bims template along with the other templates where the columns in phenotype_long_form_bims should match. Accession and rootstock can match either accession in accession_bims or progeny_name in probeny_bims. Descritor_names in descriptor_bims can be used as data in the trait column.
SNP genotype template
SNP genotype template sookSNP genotype template
As with the phenotype data, there are two templates that can be used to enter SNP genotype data. Two templates are the same except that marker names are entered as column headings in genotype_snp_wide_form_bims and as data in the marker_name column in genotype_snp_long_form_bims. As a result, there will be one row per each accession in genotype_snp_wide_form_bims and there will be multiple rows per each accession in genotype_snp_long_form_bims as shown in the diagram below. As shown below, the marker names in genotype_snp_wide_form_bims should have a $ prefix.
| *dataset_name | *Accession | *genus | *species | *marker_name | genotype | evaluator | institutional_name | sample_name |
| DC_2015_dataset | Grendadier | Malus | x domestica | AFL1 | T|G | G001 | ||
| DC_2015_dataset | Beacon | Malus | x domestica | AFL1 | G|G | B0001 | ||
| DC_2015_dataset | Grendadier | Malus | x domestica | AFL2 | T|T | G001 | ||
| DC_2015_dataset | Beacon | Malus | x domestica | AFL2 | T|C | B0001 | ||
| DC_2015_dataset | Grendadier | Malus | x domestica | GDsnp00002 | T|G | G001 | ||
| DC_2015_dataset | Beacon | Malus | x domestica | GDsnp00002 | T|T | B0001 |
| *dataset_name | *Accession | *genus | *species | $AFL1 | $AFL2 | $GDsnp00002 |
| DC_2015_dataset | Grendadier | Malus | x domestica | T|G | T|T | T|G |
| DC_2015_dataset | Beacon | Malus | x domestica | G|G | T|C | T|T |
Below are descriptions for each of the columns in genotype_snp_wide_form_bims. Columns with * are required.
- * dataset_name: name of the phenotyping dataset. It should match a 'dataset_name' column entry in the dataset_bims.
- * accession: ID of the accession that has been phenotyped. It should match an 'accession' column entry in accession_bims or ‘progeny_name' column entry in progeny_bims.
- * genus: genus to which the accession belongs to.
- * species: species name. Enter 'sp.' to represent one unknown species, 'spp.' to represent multiple unknown species.
- $ marker_name: The name of the marker is entered as a column heading with ‘$’ as a prefix. It should match the ‘marker_name’ column entry in marker_bims.
- The alleles are entered in the cell with '|' in between (e.g. A|T)
- empty cell or '-' means missing data
- '$' means null, some people use '$$' for confirmed null (confirmed using parental data), and some just use 'null' instead of '$'
- A missing value means we don't know what the target nucleotide flanking the probe sequence is whereas a null allele means that the probe sequence is either absent in the genome or that it is so different that the probe does not bind properly. Thus, a missing value represents data not available whereas a null allele is an actual value.
- The alleles are entered in the cell with '|' in between (e.g. A|T)
- evaluator: Person who did the genotyping. Multiple persons can be entered with ';' in between. It should match the contact_name'of the contact_bims.
Diagram below shows the diagram of genotype_snp_wide_form_bims along with the other templates where the columns in genotype_snp_wide_form_bims should match. Accession can match either accession in accession_bims or progeny_name in probeny_bims. The data in the marker_name column in marker_bims can be used as column headings in genotype_snp_wide_form_bims with $ prefix.
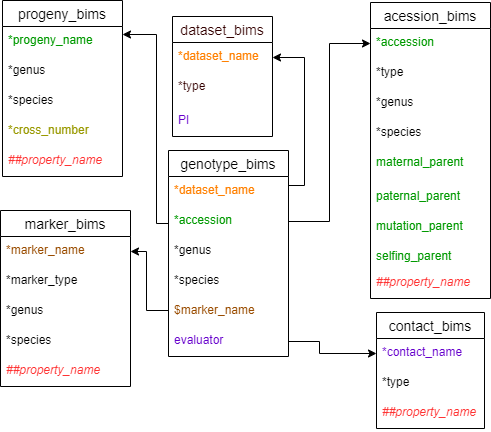
Below are descriptions for each of the columns in genotype_snp_long_form_bims. Columns with * are required.
- * dataset_name: name of the genotyping dataset. It should match a 'dataset_name' column entry in the dataset_bims.
- * accession: ID of the accession that has been phenotyped. It should match an 'accession' column entry in accession_bims or ‘progeny_name' column entry in progeny_bims.
- * genus: genus to which the accession belongs to.
- * species: species name. Enter 'sp.' to represent one unknown species, 'spp.' to represent multiple unknown species.
- * marker_name: The name of the marker. It should match the ‘marker_name’ column entry in marker_bims.
- genotype: Type the alleles with '|' in between (e.g. A|T)
- empty cell or '-' means missing data
- '$' means null, some people use '$$' for confirmed null (confirmed using parental data), and some just use 'null' instead of '$'
- A missing value means we don't know what the target nucleotide flanking the probe sequence is whereas a null allele means that the probe sequence are either absent in the genome or that it is so different that the probe does not bind properly. Thus, a missing value represents data not available whereas a null allele is an actual value.
- evaluator: Person who did the genotyping. Multiple persons can be entered with ';' in between. It should match the contact_name'of the contact_bims template.
Diagram below shows the diagram of the genotype_snp_long_form_bims along with the other templates where the columns in genotype_snp_long_form_bims should match. Accession can match either accession in accession_bims or progeny_name in probeny_bims.
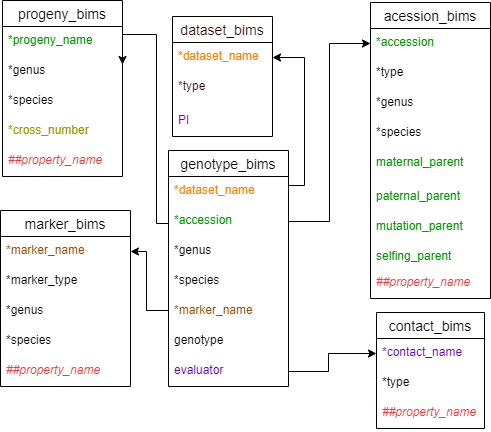
Image Templates
Image Templates sookImage Templates
There are four templates where the metadata for image files can be entered depending on the types of images. Types of images are explained in 'Importing Image Data' page. These templates need to be loaded before the actual zipped image files are loaded. If the image files are sent by BrAPI from Field Book app, the templates do not need to be loaded. Details of the columns for each of the templates can be found in Data Import-Excel Data Templates-Template list. Just click 'view' in Details column of the table.
- sample_image_bims
- accession_image_bims
- descriptor_image_bims
- marker_image_bims
Marker template
Marker template sookMarker template
The marker_bims template is used to enter marker data. Data for markers of any type can be entered.
Below are descriptions for each of the columns in marker_bims. Columns with * are required.
- dataset_name: name of the genotyping dataset. It should match a 'dataset_name' column entry in the dataset_bims template. This is used only when users need to keep the record of the dataset name for which a particular marker name was used.
- * marker_name: name of the marker.
- * marker_type: marker type such as SSR and SNP.
- * genus: The genus name of the organism from which the marker was developed.
- * species: The species name of the organism from which the marker was developed. Enter 'sp.' to represent one unknown species, 'spp.' to represent multiple unknown species.
- Alias: other names used for the marker. Multiple aliases, separated by a semicolon (;), can be added.
- SS_ID: the submitted SNPs (SS) number given by NCBI.
- RS_ID: the refSNP (RS) number given by NCBI.
- comments: Any comments about the marker.
- ##property_name: user specified custom columns that should have ## as a prefix. It should match an entry in propery_bims with the type specified as marker
- ###property_name: custom properties that are associated with markers in the specific dataset can be entered by three pound signs (###)
Match marker template
Match marker template sookMatch marker template
The match_marker_bims template is used to merge marker information in the community database and the private database in BIMS. Users can enter the marker names in the community database and the matching marker names in their BIMS program to access other information on the markers such as genome positions and primer sequences stored in the community database.
Below are descriptions for each of the columns in match_marker_bims. Columns with * are required.
- * BIMS: Marker name in BIMS.
- * Chado: Marker name in Chado (community database).